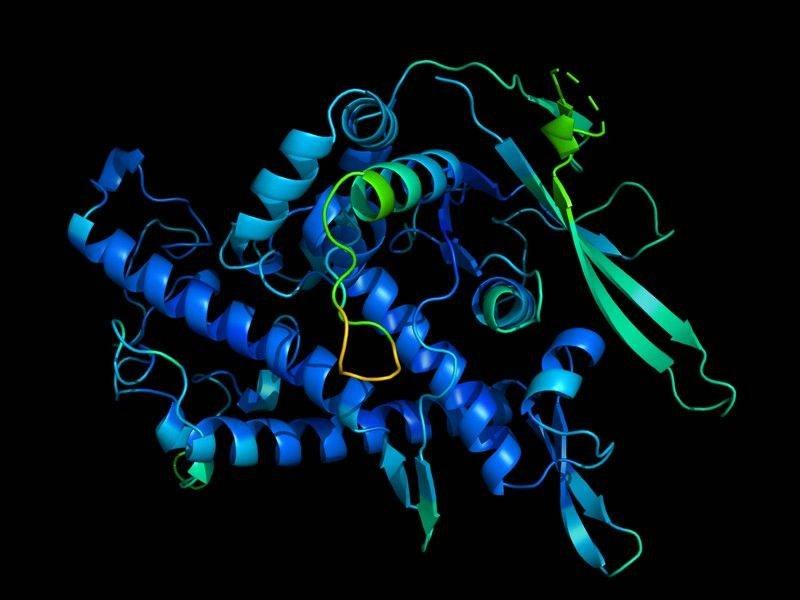
On Monday, DeepMind Technologies announced a major breakthrough in what is known in computational biology as the “protein folding problem.” Typically staid scientific communities are reacting to the news with intense excitement.
KEY POINTS
- DeepMind Technologies, a UK-based subsidiary of Alphabet since 2015, announced that it had solved a significant challenge in computational biology, the protein folding problem.
- DeepMind was previously famed for its breakthrough artificial intelligence work in games like Go. Its program “AlphaGo” was the first AI system to beat a professional Go player, Lee Sedol. The style of the AI system was surprisingly creative, not unlike human players. The match was the subject of a popular documentary eponymously titled AlphaGo.
- The company has focused on reinforcement learning, which trains computer agents to maximize their performance in complex, competitive environments with minimal inputs.
- DeepMind’s discovery will have wide-ranging impacts on molecular biology, with near term impacts most keenly felt in medicine and the pharmaceutical industry.
BACKGROUND: What is DeepMind?
DeepMind, led by Demis Hassabis and Shane Legg, is a UK-based AI firm focused primarily on reinforcement learning. This field is often considered a frontier within machine learning. Unlike other types of machine learning that rely on reams of human-annotated data, reinforcement learning trains computer systems to respond optimally to unstructured environments. In the case of DeepMind, their early work focused on developing AI systems that can effectively play simple computer games – but without any prior training or instruction.
In subsequent years, DeepMind famously developed “AlphaGo,” the first computer program to beat a professional player in the ancient game of Go. Considered by experts in artificial intelligence to be vastly more complicated than chess, the win is widely viewed as a historic milestone.
Underlying many of these advances is a broad class of algorithms and techniques often described as “deep learning.” The “deep” in “deep learning” refers to the practice of stacking many layers of simulated neurons together in an “artificial neural network.” The field of artificial neural networks was developed decades ago but was considered to be at a relative standstill until modern computing resources enabled a renaissance. The current resurgence has been pioneered by researcher Geoffrey Hinton of the University of Toronto.
Hinton’s work on deep learning sparked a revolution in the field of machine learning, ultimately leading to breakthrough advances in computer vision, and more recently, in natural language modeling.
In 2020, DeepMind has developed new deep learning methods that have enabled an extremely significant computational biology advancement.
Here is how DeepMind described the problem:
“In his acceptance speech for the 1972 Nobel Prize in Chemistry, Christian Anfinsen famously postulated that, in theory, a protein’s amino acid sequence should fully determine its structure. This hypothesis sparked a five-decade quest to computationally predict a protein’s 3D structure based solely on its 1D amino acid sequence as a complementary alternative to these expensive and time consuming experimental methods. However, a significant challenge is that the number of ways a protein could theoretically fold before settling into its final 3D structure is astronomical. In 1969 Cyrus Levinthal noted that it would take longer than the age of the known universe to enumerate all possible configurations of a typical protein by brute force calculation – Levinthal estimated 10^300 possible conformations for a typical protein. Yet, in nature, proteins fold spontaneously, some within milliseconds – a dichotomy sometimes referred to as Levinthal’s paradox.”
In short, the protein folding problem is to predict the ultimate 3D structure of a protein – a massively important aspect of its biological function – from an amino acid sequence. Predicting a protein structure without expensive experimentation has long been considered a holy grail of computational biology. In turn, predicting the 3D structure of a protein is foundational to molecular biology itself.
A prevalent pattern in machine learning research is to measure progress on significant problems through open competitions. The touchstone competition for protein folding is Critical Assessment of protein Structure Prediction (CASP). It works through a blind process where teams are sent about a hundred target proteins over several months. Each team has a period to send back predictions; then, each prediction is assessed in terms of its accuracy against the experimentally determined structure.
This plot from DeepMind shows its performance in successive years of CASP:

Any performance over 90% is considered to be roughly equivalent to experimental results. It appears DeepMind has now crossed that threshold.
According to Science, the organizers of CASP found DeepMind’s results so good that they were somewhat concerned about the possibility of cheating. To test DeepMind, they sent them an obscure target that has eluded experimental approaches. AlphaFold2 returned results that quickly led to the resolution of the problem – proving in the process that they could not possibly be cheating. The DeepMind team recounted the reaction of one of the scientists involved in the “test”:
“AlphaFold’s astonishingly accurate models have allowed us to solve a protein structure we were stuck on for close to a decade, relaunching our effort to understand how signals are transmitted across cell membranes,” stated Professor Andrei Lupas, Director of the Max Planck Institute for Developmental Biology and a CASP assessor.
ANALYSIS: Why is DeepMind On The Cutting Edge?
This development will have extremely significant impacts throughout computational and molecular biology. It will undoubtedly include medicine and pharmaceutical science. The ability to quickly predict protein structure will accelerate many research endeavors.
AlphaFold2 is particularly important in light of the earlier revolution in genome sequencing. Hundreds of millions of yet to be investigated proteins can now be much more rapidly explored using computational models. There are also massive implications for issues like the response to a pandemic, where speed in understanding a novel virus is paramount.
A case can be made that AlphaFold2 will become the most important artificial intelligence application yet produced. Among artificial intelligence and machine learning researchers, there is an ongoing fear of returning to what is sometimes called the “A.I. Winter” – a reference to a period in the 1980s when AI failed to produce meaningful advances and led to widespread pessimism and lack of funding. AlphaFold2 is potentially so significant that it could assuage such fears by demonstrating that AI can solve problems of historic magnitude.
OUTLOOK: The Future of AI
In the coming days and weeks, expect a great deal of justifiable excitement about this approach. There are several things to watch for that may slightly mitigate the result: AlphaFold still has some weaknesses with certain kinds of proteins. It will also take experts time to assess if there are any artifacts in the set of target proteins that inadvertently made the challenge easier for AlphaFold. These issues are unlikely to detract from the achievement meaningfully; still, they will provide significant indications in terms of future research.
The other factor to consider will be the computational resources required to train such complex models. According to DeepMind:
“[AlphaFold2] uses approximately 128 TPUv3 cores (roughly equivalent to ~100-200 GPUs) run over a few weeks, which is a relatively modest amount of computing in the context of most large state-of-the-art models used in machine learning today.”
These resources are certainly within reach for well-financed institutions. Expect an even greater interest in biomedical fields in investing in computational infrastructure and talent to operate similar research programs.
There will also likely be a race of sorts within industry to find ways to adopt these approaches and convert them into operational advantages. There will be new questions about business models and perhaps even monopolies: will a few AI-leaders become centralized providers of the best protein structure models? Or is there hope yet of a more democratized approach?
Few of these questions will be resolved quickly. However, they are almost sure to have a significant impact in the years to come.